UBDB-Buffalo Aspherical Atom Databank
Relativistic Scattering Factors
Relativistic Scattering Factors for Ions
Analytical Fit to Relativistic Scattering Factors
The databank contains all atom-types present in natural amino acid residues and other biologically relevant molecules. Each atom-type results from averaging over a family of the theoretical density of chemically unique pseudoatoms, taking into account both first and second neighbors. A spawning procedure is used to assure that close transferability is obeyed. The databank parameters will be listed on this website in the near future. Examples of the deformation densities of aspherical nitrogen atoms are shown below:
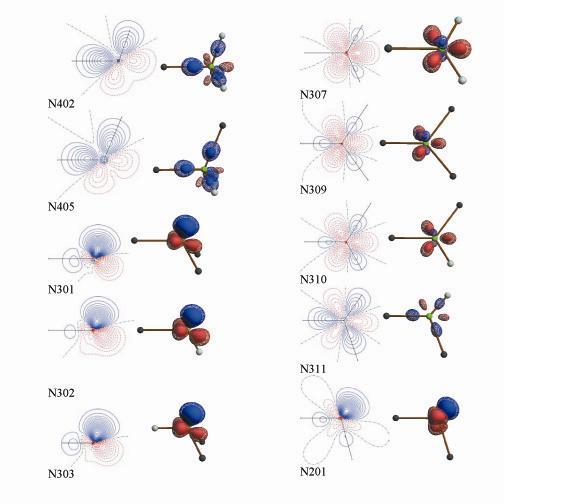
Deformation densities of the nitrogen atom types
in the UBDB: (left) in the plane of two neighboring atoms, contour
levels at 0.05 e/Å3; (right) 3D representation, contour
levels at ±0.2 and ±0.3 e/Å3. Positive contours, blue,
negative contours, red, zero contour, black.
Download University at Buffalo Pseudoatom Databank (UBDB) :
Use of UBDB implies that the user will adhere to the following terms and conditions:
- The use of UBDB should be acknowledged in any publications, which use results obtained with UBDB
Volkov, A., Li, X.,
Koritsanszky, T. S. & Coppens, P.,
J. Phys. Chem. A
108, 4283-4300, (2004).
Dominiak, P. M.,
Volkov, A., Li, X., Messerschmidt, M. & Coppens, P., J. Chem. Theory
Comput., 3,
232-247
(2007).
Volkov, A.,
Messerschmidt, M. & Coppens, P., Acta Crystallogr. D
63, 160-170, (2007).
Download
databank in WINDOWS format
Download databank
in UNIX format
Download program LSDB :
Use of the program LSDB implies that the user will adhere to the following terms and conditions:
-
The use of the program of LSDB should be acknowledged in any publications, which use results obtained with LSDB
-
The acknowledgment should be of the form:
Volkov, A., Li, X.,
Koritsanszky, T. S. & Coppens, P.,
J. Phys. Chem. A
108, 4283-4300 (2004).
Dominiak, P. M.,
Volkov, A., Li, X., Messerschmidt, M. & Coppens, P., J. Chem. Theory
Comput., 3,
232-247
(2007).
Volkov, A.,
Messerschmidt, M. & Coppens, P., Acta Crystallogr. D
63, 160-170, (2007).
Download statically linked x86
LINUX executable of the program LSDB
Download WINDOWS
executable of the program LSDB
LSDB usage notes:
1. LSDB requires input file called lsdb.inp
2. Running LSDB executable with '-h' option will printout the sample lsdb.inp file with
explanation of each of the options (see below)
3. Download and run example (files are in WINDOWS format)
4. Sample
lsdb.inp file :
<>!
! Example of LSDB input
file (lsdb.inp) :
!
FORMAT *shelx xd
aim2tab platon
! for SHELX :
INPUT
l-dopa.shelx rename 0 toxd
! for XD :
!INPUT xd.mas
xd.inp
! etc :
!INPUT file.inp
! printout level (higher
number gives more printout) :
VERBOSE 4
! tolerances for bonds /
planar rings / planar groups
TOLS bond 0.4 ring 0.1
group 0.1
! modify covalent radii
!RADII H 0.1
He 0.2 Li 0.3 ....
! number of cells in each
direction for complete fragment search
! comment it out or set
to -1 to turn off search
NCELLS 1
! how to handle
disordered part
DISORD
extendH db
! generation of keys
KEYGEN XYZUIJ all
ordered MULT all ordered
! use simple model for
determination of chemical equivalency
SIMPLE
! use/do_not_use
chemical/kappa contraints for H/non-H atoms
CHEMCON
*hydrogen *other
! extend H-atoms to
neutron/standard distance
Extend_H
! search for 3-8 membered
rings
RINGS 3
8 *special
! assign pseudoatom
parameters from COPPENS LAB databank
DATABANK
/usr/local/UBDB_Oct10_2006_unix.db
! select method for
scaling of Pv's
SCALE *sigma diff fract
! number of fragments
NFRAG 1
! fragment specifications
!FRAG 1 1 -25
!FRAG 2 26 -50
! net charges of fragments
!CHFRAG 0. 0.