When
integrating an atomic basin TOPXD gives the following error message:
PATHE2: OSCILLATION OF PATHS
PATHE2: THE ATTRACTOR OF THIS PATH WAS PROBABLY NOT INCLUDED IN THE CLUSTER
Did you check the list of atoms reached into the feeler rays determination
step?
Do you think that some neighbouring atoms were missed? If so, you
have to increase NVI parameter in order to include the missing atoms
into the list of possible attractors of trajectories of the charge density.
The OSCILLATION OF
PATHS message may also appear in some cases where the
integration will be anyhow successful. In many instances it represents
just a warning. Especially, if you noticed that the list of neighboring
atoms (after the feeler ray step) corresponds to your expectations.
Check the list of atoms reached in the feeler ray step. Increase NVI
parameter,
if needed, and then start again. Once you have used a very large NVI
value, leave your calculation to try to end its task (even if the message
appear many times).
In order to obtain satisfactory results you should use something like:
64*48*120 (phi*theta*radial) for second-row
atoms
32*24*96 (phi*theta*radial) for hydrogen atoms (if not involved in H-bond)
48*32*96 (phi*theta*radial)for hydrogen atoms (if involved in H-bond)
32*24*96 (phi*theta*radial) for hydrogen atoms (if not involved in H-bond)
48*32*96 (phi*theta*radial)for hydrogen atoms (if involved in H-bond)
Computing time is roughly proportional to n(phi)*n(theta)
points. The number of radial points is very important for the precision,
but hardly affects the total integration time, as it is operative only
in the integration step and NOT in the ZFS determination (which takes about
95% of the total time).
Unfortunately the integration step is very very long (especially the ZFS
determination which takes about 95% of this time). You can try with the
other proposed method (which is much faster), but it often fails.
Using the indirect method (the one you are using) you can save some time
by decreasing the accuracy of the surface determination. It is 0.001
(see parameter ACCUR in subroutine TOPON)
One could try to increase it up to 0.003 (no more than 0.005,
I would say). You loose somewhat in precision, but you certainly increase
in speed.
You could compare the results of these two computations on one of the atoms
you have already integrated, 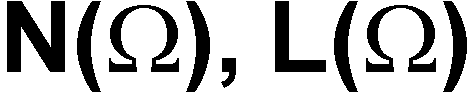 ,
etc. using:
,
etc. using:
a) first test : 64*48*120 ACCUR=0.001
b) second test : 64*48*120 ACCUR=0.003
Then you can decide if it is worth varying such a parameter and how much
you can vary it.
How
do I check the accuracy of the integration ?
Check the value of the integrated Lagrangian. For "exact" integration it
should vanish (for the divergence theorem).
In practice :
a) It should be less than 5*10-5 for H atoms, possibly around 1*10-5. A value of 1*10-4 could be perhaps acceptable, but not too precise.
a) It should be less than 5*10-5 for H atoms, possibly around 1*10-5. A value of 1*10-4 could be perhaps acceptable, but not too precise.
b) For second row atoms (C,N,O, etc,) it should not exceed 1*10-3.
Possibly 1*10-4
It will affect it, but in a very limited way, especially after the feeler
ray step. Indeed the atoms reached during the feeler ray step are put at
the top of the list of the NVI reachable atoms. So that the
DO
loop in PATHEN and PATHEN2 (these DO run on the 3*NVI*STAR_MULTIPLICITY
coordinates of the possible attractors) are in most cases (>99%)
terminated much before the end of the loop.
In practice you shouldn't notice a CPU time increase with NVI increase.
Rather you could notice a decrease, if you have added an attractor that
had to be enclosed. In this case the path oscillation is avoided and CPU
time considerably saved.
When
trying to increase the number of points to something like 128*96, TOPXD
gives the following error message:
ERROR
**** YIELD**** GAUSS QUADRATURE NOT AVAILABLE - NUMBER OF POINTS= 128
Actually the current programs works with these number of points for
the quadrature:
1, 2, 4, 6, 8, 10, 12, 14, 16, 18, 20, 22, 24, 26, 28, 30, 32, 36, 40,
48, 64, 96, 120
So 128 it is not a valid option. 120*96 should work.
What about electroneutrality? Are you very far from it ?
The fact that one may get problems with carbon or nitrogens and never with
oxygens or hydrogen atoms seems to indicate that the former have more complicate
ZFS's than the latter. You could try to solve such a problem, by increasing
the radius of the beta sphere for such atoms, thus reducing the size of
the remaining part of the atomic basin.
You could use for the beta sphere something like the distance of the closest
bond critical point multiplied by 1.15 (the program then reduces this number
by 20%). Furthermore, the increase (inside the code) of the number of theta
and phi points in the inner beta sphere might help. Please contact us and
we will send you instructions on how to do it